CRO事業
CROとは
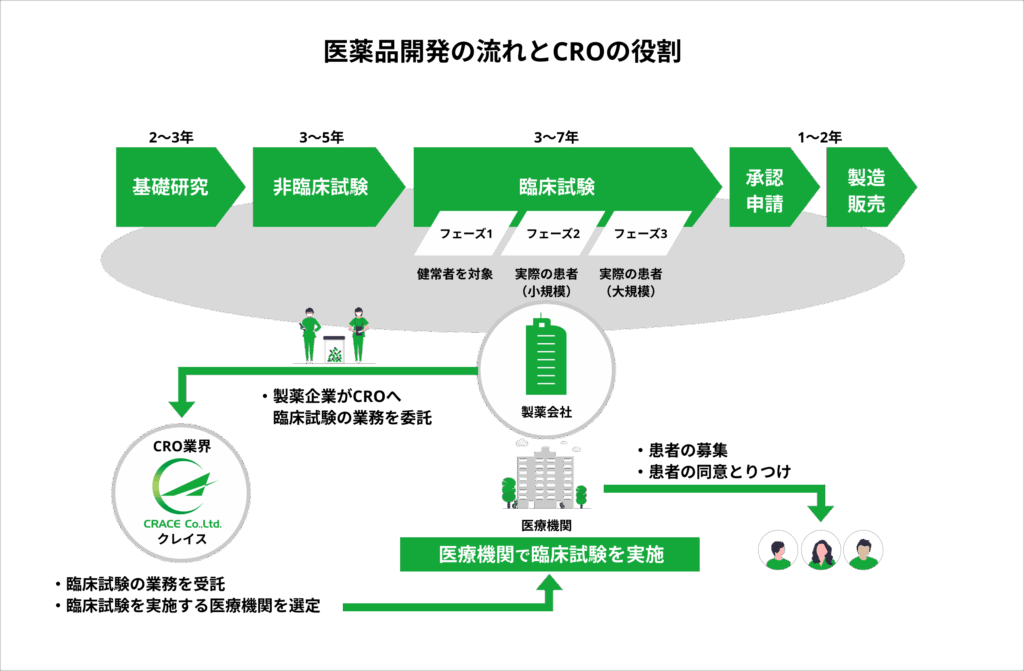
CRO(Contract Research Organization:医薬品開発業務受託機関)とは、製薬企業やバイオベンチャーなどの治験依頼者から委託を受け、治験の計画立案、モニタリング、データ管理、統計解析など医薬品や医療機器の開発に関わる多岐にわたる業務を支援・代行する専門機関です。
医薬品開発の流れとCROの役割
開発受託サービス
国内外の企業治験・医師主導治験ともに、経験豊富なメンバーが高品質なサービスを提供いたします。フルサポートはもちろん、各スポット毎のご依頼などお客様のニーズに合わせて柔軟に対応いたします。また、臨床開発のエキスパートとケアネットプラットホームを掛け合わせることで、試験全体のリードタイムを短縮いたします。
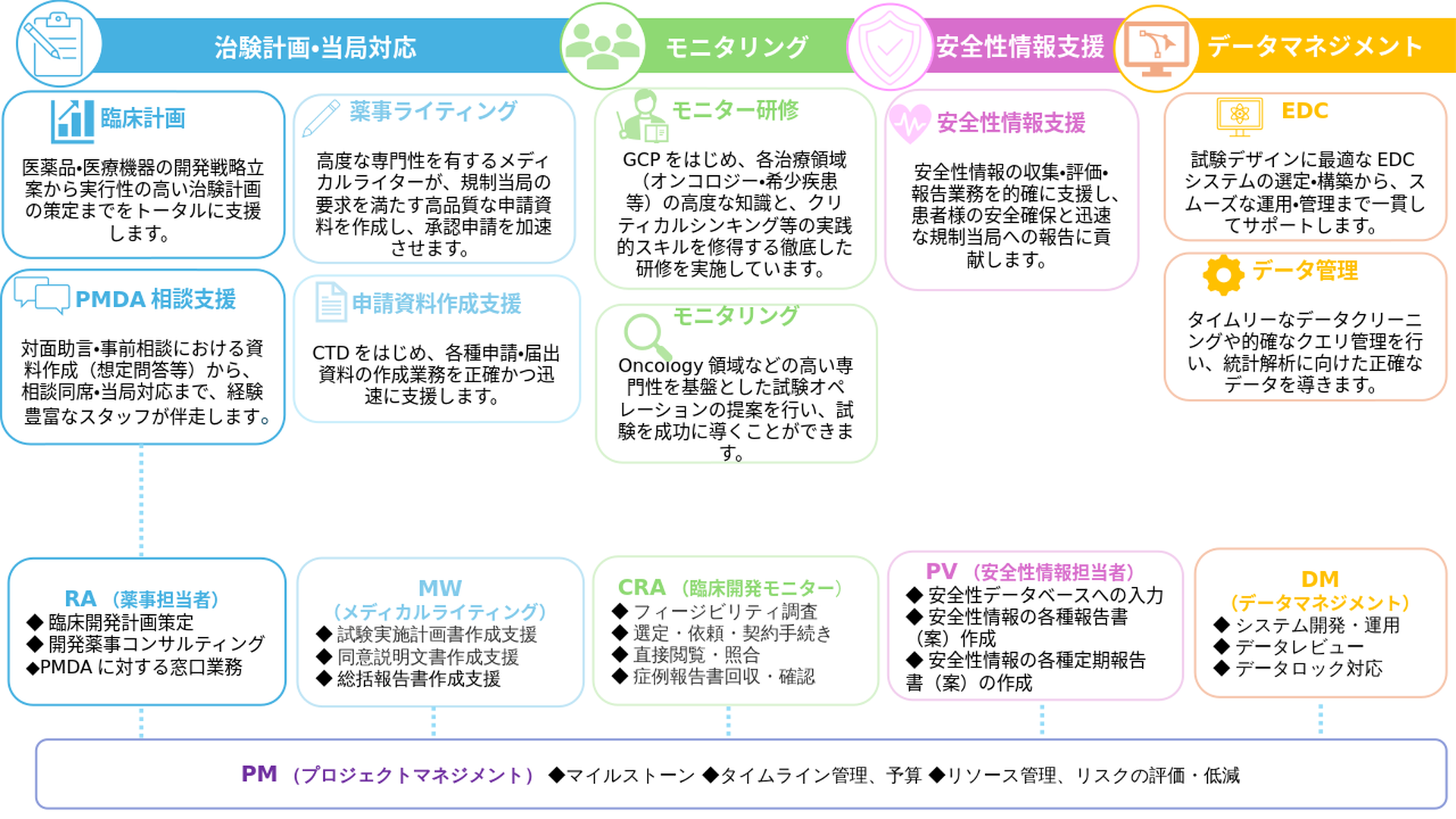
 治験計画・当局対応
治験計画・当局対応
 モニタリング
モニタリング
 安全性情報支援
安全性情報支援
 データマネジメント
データマネジメント
 臨床計画
臨床計画医薬品・医療機器の開発戦略立案から実行性の高い治験計画の策定までをトータルに支援します。
 薬事ライティング
薬事ライティング高度な専門性を有するメディカルライターが、規制当局の要求を満たす高品質な申請資料を作成し、承認申請を加速させます。
 PMDA 相談支援
PMDA 相談支援対面助言・事前相談における資料作成(想定問答等)から、相談同席・当局対応まで、経験豊富なスタッフが伴走します。
 申請資料作成支援
申請資料作成支援CTDをはじめ、各種申請・届出資料の作成業務を正確かつ迅速に支援します。
 モニター研修
モニター研修GCPをはじめ、各治療領域(オンコロジー・希少疾患等)の高度な知識と、クリティカルシンキング等の実践的スキルを修得する徹底した研修を実施しています。
 モニタリング
モニタリングOncology 領域などの高い専門性を基盤とした試験オペレーションの提案を行い、試験を成功に導くことができます。
 安全性情報支援
安全性情報支援安全性情報の収集・評価・報告業務を的確に支援し、患者様の安全確保と迅速な規制当局への報告に貢献します。
 EDC
EDC試験デザインに最適なEDCシステムの選定・構築から、スムーズな運用・管理まで一貫してサポートします。
 データ管理
データ管理タイムリーなデータクリーニングや的確なクエリ管理を行い、統計解析に向けた正確なデータを導きます。
- 臨床開発計画策定
- 開発薬事コンサルティング
- PMDAに対する窓口業務
- 試験実施計画書作成支援
- 同意説明文書作成支援
- 総括報告書作成支援
- フィージビリティ調査
- 選定・依頼・契約手続き
- 直接閲覧・照合
- 症例報告書回収・確認
- 安全性データベースへの入力
- 安全性情報の各種報告書(案)作成
- 安全性情報の各種定期報告書(案)の作成
- システム開発・運用
- データレビュー
- データロック対応
•PM(プロジェクトマネジメント)…プロジェクト全体の指揮を執り、タイムラインと品質を徹底管理
治験の立ち上げから完了に至るまで、プロジェクト全体のスケジュール、コスト、品質、そしてリスクを統括します。クライアント企業様のパートナーとして、各専門部署(CRA・DM等)を連携させ、開発戦略の確実かつ迅速な実行をリードします。
•RA(薬事担当者)…最新の規制をクリアし、承認申請への最短ルートを導く
国内外の医薬品規制(GCP、各種ガイドライン)に精通したスペシャリストが、実効性の高い開発計画の策定をサポートします。当局(PMDA等)への各種届出・申請書類の作成から、戦略的な照会事項への対応まで、コンサルティングを含めた多角的なアプローチで早期の上市(承認取得)へと導きます。
•MW(メディカルライティング)…高度な科学的知見を、正確かつ論理的な「文書」へ昇華させる
治験実施計画書、総括報告書、承認申請資料(CTD)など、医薬品開発に関わる多種多様な科学的ドキュメント作成を支援します。高い倫理観と科学的客観性に基づき、審査に耐えうる高精度な文書を提供することで承認申請を加速させます。
•CRA(臨床開発モニター)…医療機関と強固な信頼を築き、治験の「質」と「安全性」を守る
治験が実施される医療機関(病院など)の選定から、契約手続き、進捗管理、モニタリング業務までを担います。GCP省令や治験実施計画書が厳格に守られているかを現場で確認し、医療機関との綿密なコミュニケーションで、新薬の承認申請に必要不可欠な「臨床データ」の信頼性を担保します。
•PV(安全性情報担当者)…医薬品の安全性を見守り、リスクを最小限に抑える
国内外から寄せられる膨大な安全性情報を迅速かつ正確に収集・評価し、当局への報告業務を的確にご支援いたします。科学的な視点に立ったきめ細やかな安全管理により、患者様の安全を守るサポートをいたします。
•DM(データマネジメント)…膨大な臨床データを、価値ある「信頼のデータ」へ構築する
治験で収集された膨大な症例データを、統計解析や承認申請に耐えうる「クリーンなデータ」へと整えます。EDC(電子症例報告書)の構築からデータチェック、ロジカルチェックなど、データ固定に至るまで全プロセスの品質を厳格に管理し、治験の科学的根拠を底上げします。
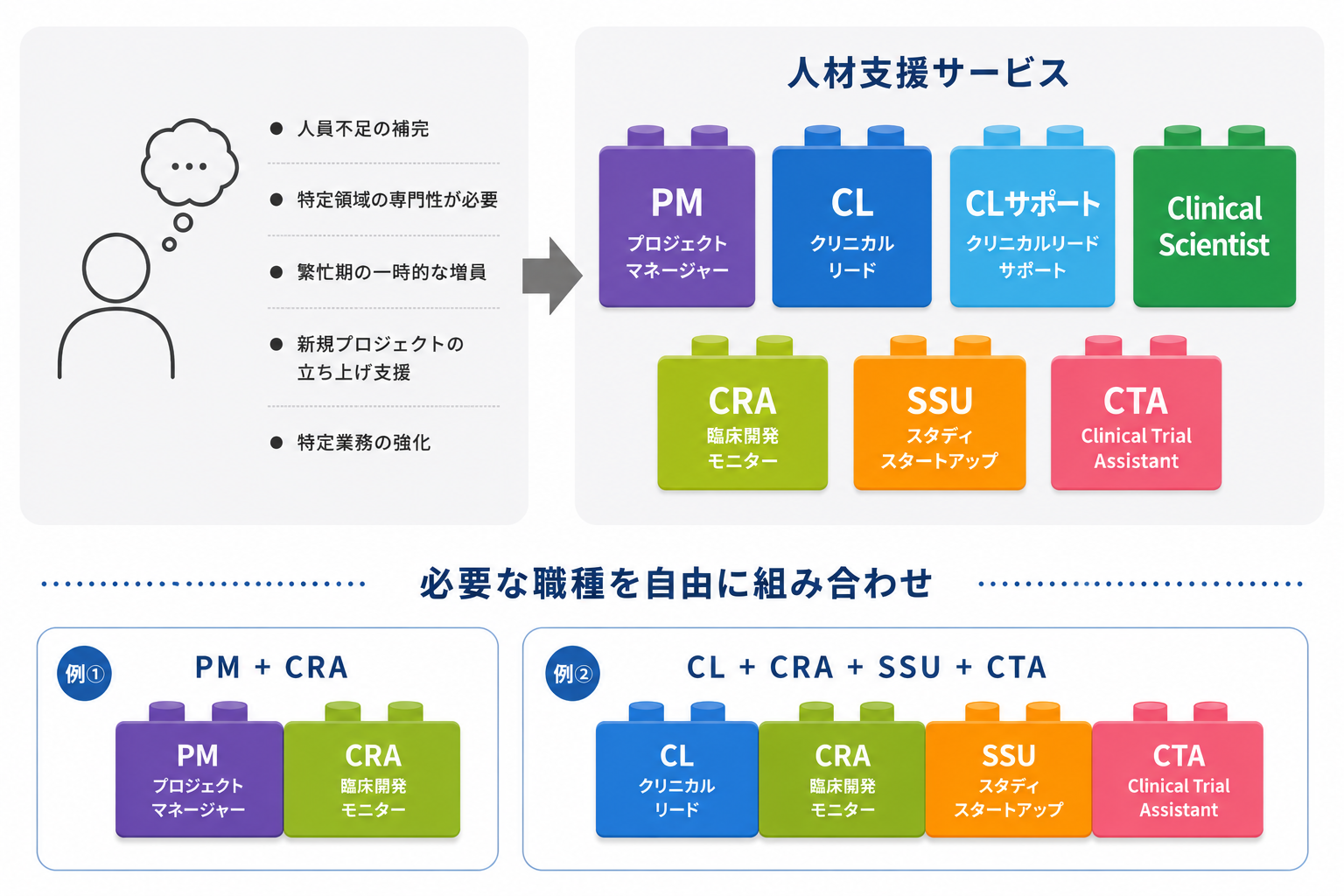
人材支援サービス
各領域の高度なスペシャリストを即戦力として派遣・提供いたします。試験の状況や課題に応じて、最適なリソースを「チーム体制」でも「スポット(単一職種)」でも、”必要な期間、必要な分だけ” 柔軟に提供いたします。
•PM(プロジェクトマネジメント)
•CL(クリニカルリード)
•CLサポート(クリニカルリードサポート)
•Clinical Scientist
•CRA(臨床開発モニター)
•SSU(スタディスタートアップ)
•CTA(Clinical Trial Assistant)